电催化CO2还原反应(eCO2RR)是将CO2转化为更有价值的化学产品的一种方法,也是一个解决全球性碳排放过量和化石能源短缺的极有前景的策略。目前研究发现,铜基的电催化剂可将CO2还原为具有更高价值的多碳产物,然而,在eCO2RR过程中,铜基催化剂会经历动态结构演变,导致难以建立稳定可靠的构效关系,极大阻碍了铜基催化剂的进一步发展和设计。因此,深入理解铜基催化剂在工况下动态结构演变的微观机理,对于催化剂的优化设计、有针对性的调控反应速率和选择性,以及加速Cu基eCO2RR催化剂实际应用具有重要的指导意义。
近日,西安交通大学化学学院丁书江团队苏亚琼教授课题组与华中科技大学夏宝玉教授课题组基于恒电位的第一性原理计算方法和多种实验表征手段,以Cu-N-C催化剂为研究模型,深入探究并提出了Cu单原子的动态烧结机理,并揭示了不同催化剂构型与eCO2RR性能之间的联系以及催化剂上C2产物的活性来源,同时就如何进一步控制反应中催化剂构型等重要问题进行了讨论。
研究团队以Cu-N-C单原子催化剂为研究模型开展了深入研究。系统性的恒电位第一性原理分子动力学(AIMD)模拟结果揭示,H和eCO2RR中间体(例如,CO)在Cu-N-C上的协同吸附可促进Cu单原子通过形成Cu-(CO)x复合物来发生脱嵌,并在随后形成团簇。而烧结形成的Cu团簇上可以快速的发生CO-CO偶联,这对于进一步生成C2产物起着关键作用(机理示意图如下图所示),提高Cu2+浓度或外加电位可以抑制Cu的这一烧结过程。
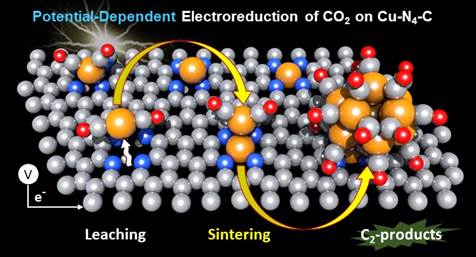
电催化CO2还原过程中Cu‒N‒C催化剂上CO中间体吸附辅助的Cu动态烧结机理示意图
研究团队计算了在298.15K时,pH-电位的坐标空间中,Cu2+离子浓度分别为10-9 M、10-6 M、10-3 M和1 M时CuN4位点抵抗Cu2+溶解的热力学稳定性结果,如下图所示,橙色区域代表了CuN4处于热力学稳定的区域。结果表明,即使在Cu2+浓度较高(1 M)的环境中,Cu2+的溶解在酸性或中性条件下(溶液pH < 7.3)仍然在热力学上是放热的,而在pH ≥ 7.3时,若外加电位显著更负于-1.26 VSHE的也可能引起Cu2+的溶解。而在常见的Cu2+离子浓度下,如10-6 M或10-3 M,将电解质pH提升至超过10.3,并同时调整应用电位至高于-1.45 VSHE,可作为抑制CuN4的Cu2+溶解的一般策略。

A) 展示了涉及五条Cu2+溶解路径的相应原子结构,其中铜、氮、碳和氢原子分别以橙色、蓝色、灰色和白色表示 B) Cu2+浓度为10-9 M C) Cu2+浓度为10-6 M D) Cu2+浓度为10-3 M E) Cu2+浓度为1 M时Cu热力学稳定性与pH和外加电位的关系
随后基于吸附能的计算结果发现,Cu-N-C中,H在N位点的吸附和CO在Cu位点的吸附存在协同作用。如下图所示。进一步研究发现,Cu位点在反应条件下可同时吸附两个CO,并且Cu单原子可进一步通过形成Cu-(CO)2复合物的形式发生脱嵌,该过程动力学势垒在-1.0 VSHE下极低。随后在反应条件下的AIMD模拟中进一步观察到了该过程的发生,并且进一步的AIMD模拟观察到了形成的Cu-(CO)2复合物经过迁移后被另一CuN4位点捕获,形成Cu-Cu键,发生初步烧结的过程。这部分计算结果有力支持了CO吸附辅助Cu单原子脱嵌烧结机理的可行性。
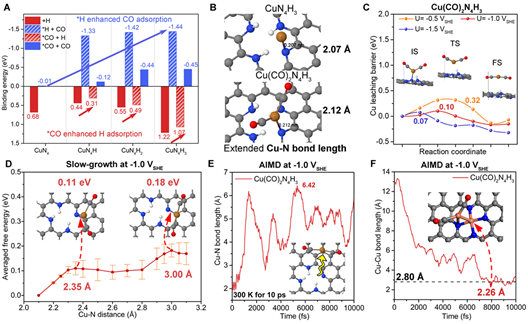
A) CO和H在不同基底上的结合能 B) CuN4H3和Cu(CO)2N4H3的优化结构
C) 在U = - 0.5、-1.0和-1.5 VSHE时Cu(CO)2N4H3上的Cu溶出势垒
D) 通过“Slow-growth”方法计算得到Cu(CO)2N4H3在-1.0 VSHE时Cu-(CO)2结构溶出过程中的总自由能结果
E) Cu(CO)2N4H3在10 ps的恒电位AIMD模拟期间的Cu-N键长 F) Cu(CO)2N4H3在额外的10 ps期间的Cu-Cu键长
对Cu团簇位点与Cu单原子位点上的eCO2RR自由能和微观动力学计算结果表明,反应条件下,TOF的最大值出现在ΔGCOOH约为1.2 eV的位置,其中CuN4和CuN4H位点在所有考虑的位点中活性最高。由于铜团簇对CO具有较强的结合强度,计算也表明其上CO覆盖度较高,因此进一步基于覆盖CO的铜团簇模型计算了CO-CO偶联的势垒,结果显示该势垒仅为0.15 eV,该位点上可快速的发生CO-CO偶联,有利于进一步的形成C2产物。这部分结果表明,实验中在Cu-N-C催化剂上观测到的C2产物主要来源于在较负电位下Cu烧结产生的Cu团簇位点。
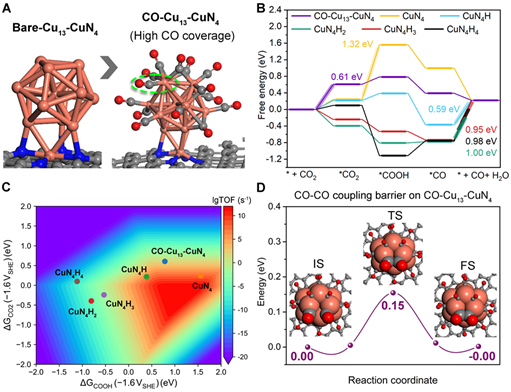
A) CO-Cu13-CuN4的优化后原子结构
B) 在CuN4、CuN4H、CuN4H2、CuN4H3、CuN4H4和CO-Cu13-CuN4上的eCO2RR自由能图
C) 在-1.6 VSHE下,六种催化位点上CO2还原生成CO的催化活性火山图
D) 计算得到的CO-Cu13-CuN4上的CO‒CO偶联动力学势垒
综上所述,研究团队发现,在eCO2RR条件下Cu-N-C催化剂上H和CO的吸附存在协同效应,导致Cu-羰基物质(如Cu-(CO)2)的形成,并显著加速Cu原子在Cu-N-C催化剂中的溢出和团聚。通过CO辅助机制,Cu溶出的动力学壁垒降低,特别是在低电位下。提高电位可以显著减少Cu-N-C催化剂上的CO和H覆盖,并显著抑制Cu烧结。而由于形成的Cu团簇可以提供极低的CO‒CO偶联壁垒,结合微观动力学分析,结果强烈表明Cu团簇可以作为生成C2产品的主要位点。进一步的实验研究证实了在电催化CO2还原反应过程中,关键的CO等eCO2RR中间体促进了Cu-N-C催化剂上Cu团簇的大量形成。本研究有助于增进对电催化CO2还原反应中Cu行为的复杂机制的理解,并对合理设计Cu基的eCO2RR单原子催化剂以产生C2+产物具有重要意义。
该研究相关成果4月8日在线发表于《德国应用化学》(Angewandte Chemie International Edition)。西安交大化学学院为论文第一单位,博士研究生秦棪阳、赵文杉、夏琛沣为共同第一作者,武甜甜博士、苏亚琼教授与夏宝玉教授为共同通讯作者。该项工作获得了国家重点研发计划、学校青年拔尖人才支持计划等多个项目资助。
文章链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202404763








