1,n-双官能化烷基连接子是构建复杂有机分子、功能材料和药物分子的重要合成砌块,在有机合成、药物化学及功能材料领域具有广泛应用价值。然而,传统制备方法主要依赖从头合成,存在步骤经济性差、官能团兼容性有限、远端双官能化(n≥4)构建困难等瓶颈。如何从廉价易得的环烷酮出发,通过简洁高效的途径实现远端双官能化,是有机合成领域长期面临的挑战。
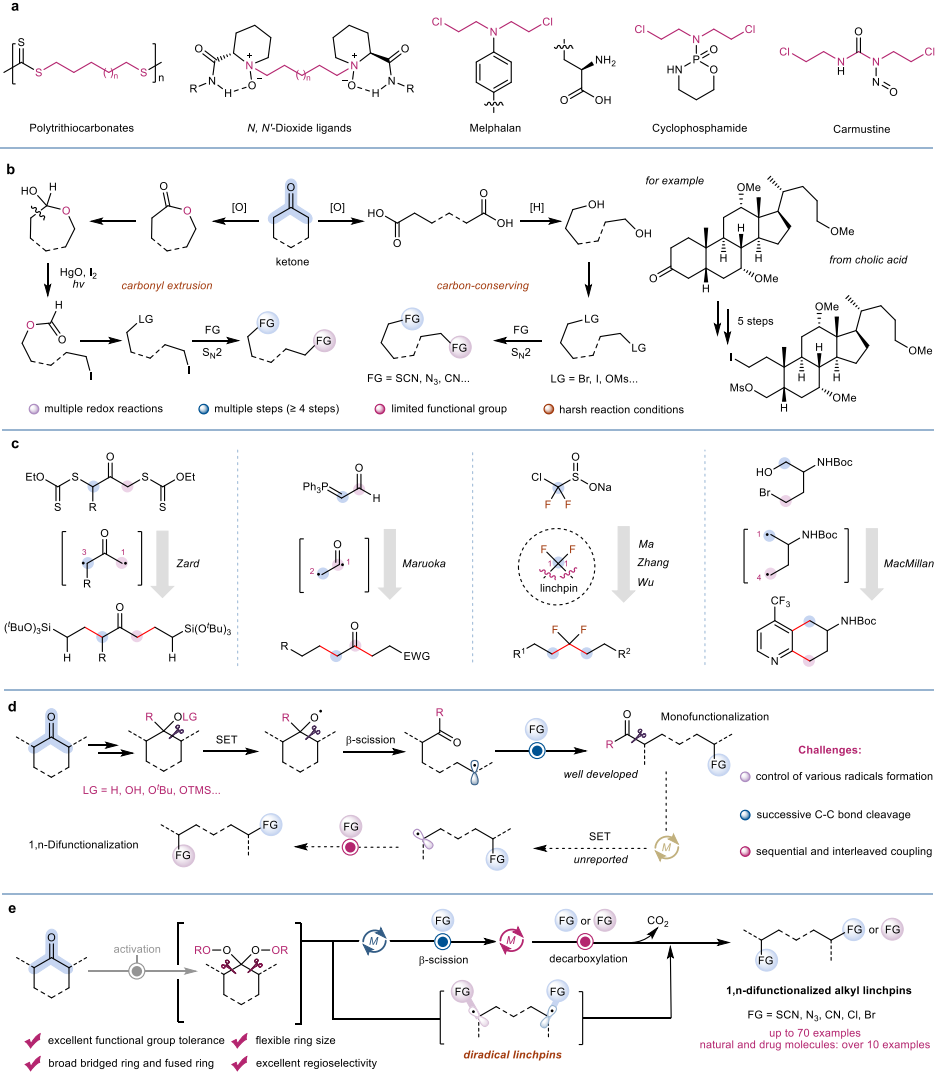
针对上述挑战,西安交通大学郭丽娜教授团队创新性地提出将环烷酮转化为偕二过氧化物,作为潜在的双自由基连接子。通过精巧设计铁(铜)催化串联过程,成功整合了烷氧自由基诱导的β-断裂与酰氧自由基介导的脱羧反应,实现了环烷酮的开环、碳链收缩及双向官能化。该策略的核心在于精准调控两种自由基中间体的生成时序与催化循环的相容性,从而完成连续可控的碳碳键断裂与官能团引入,为远端双官能化连接子的模块化合成开辟了新路径。该催化体系展现出优异的底物适用性与官能团耐受性:兼容五至十五元环、桥环、稠环等多样环系骨架,耐受芳基、烷基、酯基、磺酸酯等多种官能团。基于该策略,研究团队成功构建了结构多样的烷基1,n-二硫氰酸酯、1,n-二叠氮化物、1,n-二卤化物(n≥4),并开发了“一锅两步法”合成非对称的1,n-硫氰酸-叠氮连接子。值得关注的是,该方法还成功应用于复杂天然产物及药物分子的后期编辑——包括脯氨酸、布洛芬、薄荷醇、5-脂氧合酶抑制剂以及多种甾体酮类药物分子,均能以良好收率得到目标产物。在多羰基甾体酮底物中,反应表现出优异的区域选择性,优先在位阻较小的环酮位点发生选择性开环官能化。
机理研究表明,该反应经由烷氧自由基诱导的b-断裂生成烷基自由基,随后与Fe(III)-SCN配合物作用形成过氧酯中间体;该中间体经单电子还原生成酰氧自由基,脱羧后生成第二个烷基自由基,最终与另一分子亲核试剂偶联得到目标产物。这一串联过程采用一种催化剂实现两个催化循环(“一石二鸟”),实现了双向碳碳键断裂与官能团引入的精准衔接。产物可进一步转化为二硫醚、环状硫醚、双三氮唑、双磷酰胺等复杂分子,并通过点击化学实现与药物分子的高效连接。这些衍生化转化充分展示了该策略在分子编辑和复杂1,n-双官能化烷基分子合成中的广阔应用前景。
该研究成果以《酮类化合物的碳环编辑通过自由基介导的双向碳碳键断裂/偶联策略》(Carbocycle editing of ketones proceeds via a radical-mediated bidirectional C-C bond cleavage/coupling strategy)为题发表在国际知名学术期刊《自然·通讯》(Nature Communications)上。西安交通大学化学学院为该论文第一完成单位,博士研究生马英杰为第一作者,郭丽娜教授为论文通讯作者。该研究得到国家自然科学基金和西安交通大学AI科学专项研究基金等项目支持并获得了西安交通大学分析测试共享中心在测试表征方面的支持。
论文链接地址:https://doi.org/10.1038/s41467-026-74317-0
郭丽娜教授课题组主页:https://gr.xjtu.edu.cn/guoln81/








