作为大脑的基本功能单位,神经元依赖于神经分泌与突触传递进行相互间信息交流,而正是数以亿计的神经元所构建的神经网络中信息的交流与整合,赋予了大脑各种生理功能以调控生命活动。神经分泌的本质是由细胞内Ca2+信号精密调控“融合分子机器”-SNARE复合物的有序组装,驱动囊泡膜与细胞质膜的融合,从而完成囊泡内的化学物质(神经递质、神经肽等)从分泌囊泡释放到胞外的过程。突触结合蛋白(Synaptotagmin,Syt)作为触发神经分泌的Ca2+传感器与分子开关,通过与SNARE复合物形成“融合分子机器”,触发囊泡融合与递质分泌。然而,这些“融合分子机器”动态组装的分子结构基础及其机制尚不明确。
作为神经分泌的分子开关,Syt蛋白依据其Ca2+结合能力,可分为Ca2+亲和性Syts(CaR-Syts,如Syt1)和Ca2+非亲和性Syts(CaT-Syts, 如Syt11)。西安交通大学徐华栋/王昌河/宋茜/焦联营团队前期研究发现,Syt11作为Ca2+非亲和性Syt,不但不能触发神经递质分泌,还是胞吐后胞吞过程的重要刹车蛋白,还是确保多巴胺分泌稳态调控的重要“刹车枢纽”(Nat Commun, 2024; PNAS, 2022; Nat Commun, 2018; EMBO Rep, 2016)。然而,其抑制胞吞作用的分子基础及其机制尚不清楚。更重要的是,在突触传递过程中,CaR-Syts和CaT-Syts是否及如何协同调控兴奋-分泌偶联和胞吐-胞吞的偶联平衡也不清楚。
近日,研究团队首次发现,Syt11通过与Syt1竞争结合细胞质膜参与神经分泌的动态调控,Syt11虽为CaT-Syt典型代表,它与质膜的结合也受Ca2+严格调控:Ca2+促进CaR-Syts但抑制CaT-Syts与质膜的结合,Ca2+对磷脂膜表面产生静电屏蔽抑制CaT-Syts的结合。该研究不仅发现Ca2+对Syt-质膜互作的“促进”和“抑制”双向调控机制,阐明了CaR-Syts和CaT-Syts与质膜结合及其Ca2+敏感性的分子基础,建立了Syt蛋白竞争性“质膜占位动态切换”理论模型,揭示了神经元兴奋-分泌偶联平衡新机制,为胞吐-胞吞偶联平衡提供新视角和新理论。
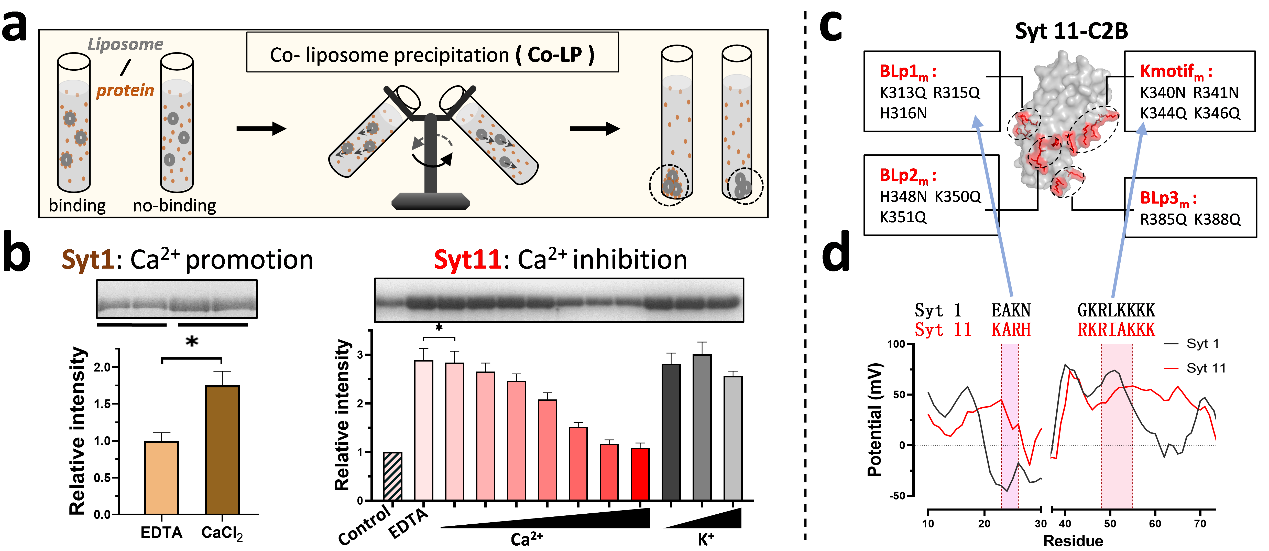
CaT-Syts的膜结合具有Ca2+敏感性
研究团队使用脂质体共沉淀(co-LP)方法分别检测CaR-Syt1与CaT-Syt11与脂质体的相互作用。两者都能在无Ca2+环境中结合脂质体;作为CaR成员,Ca2+可促进Syt1与质膜的结合;而有趣的是,作为CaT成员,Syt11与质膜的结合也表现出严格的Ca2+依赖性,可被Ca2+强烈抑制。进一步利用点突变进行筛查发现,Syt11的C2B上存在多个碱性区域,共同、冗余地保证Syt蛋白与膜的结合,其中还包括一处仅在CaT-Syts中呈碱性、而在CaR-Syts中呈酸性的独特位点。这些结果表明,Syt蛋白与磷脂膜存在广泛的不同模式的相互作用,而Ca2+对CaR与CaT两类Syts的调节作用刚好相反:促进CaR-Syts与质膜的结合,而抑制CaT-Syts与质膜的互作。
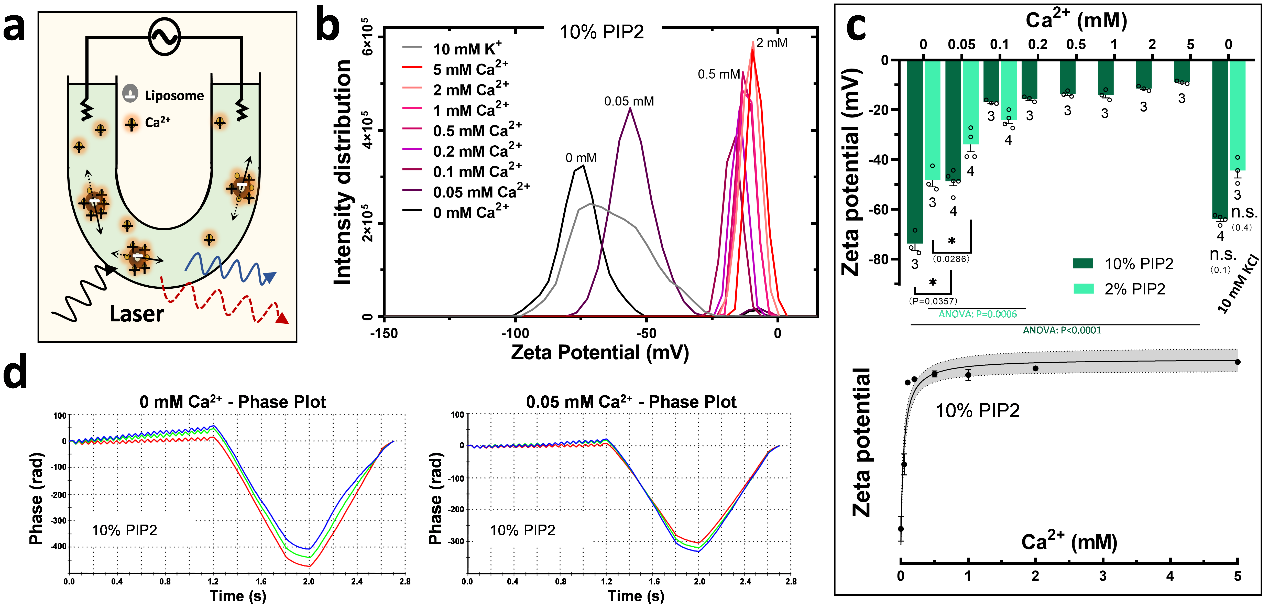
Ca2+提升脂质体表面静电势
由于CaT-Syts不具备Ca2+亲和性,为了探究Ca2+抑制其膜结合的分子机制,研究团队猜测Ca2+可能通过与膜磷脂的相互作用来抢占Syts的结合位点。利用电泳光散射(ELS)检测Ca2+对脂质体(Φ 400 nm)表面静电势的影响发现,50 μM Ca2+足以大幅度地提升脂质体的Zeta电位。这表明Ca2+可被磷脂膜组成成份PIP2头部的磷酸基团螯合,从而屏蔽磷脂膜表面的静电荷,抑制CaT-Syts与质膜的结合。
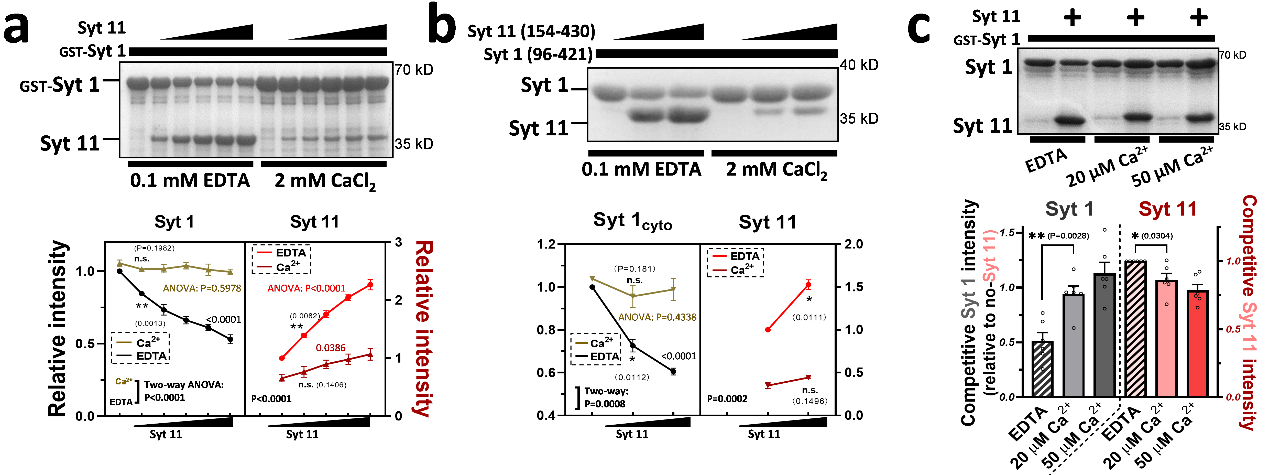
Ca2+抑制Syt11与Syt1的竞争性膜结合
考虑到Syt1与Syt11在生理条件下共存及Ca2+对两者膜结合能力的相反效应,为验证同一体系内Ca2+对两者互作关系的调节作用,研究团队通过体外实验发现,Syt11在无Ca2+环境中可凭借自身的强大膜结合能力与Syt1竞争结合脂质体膜,但Ca2+的引入会抑制Syt11并促进Syt1,从而逆转它们之间的竞争关系。对Ca2+敏感性的研究发现,生理性Ca2+水平(20 μM)足以大幅削弱此竞争效应。此结果表明,Syt11对Syt1的竞争性膜结合是Ca2+敏感的;而在正常的神经传递活动中,Syt1与Syt11之间的竞争性膜占位很可能受神经元兴奋性(胞内Ca2+浓度)的动态调控。
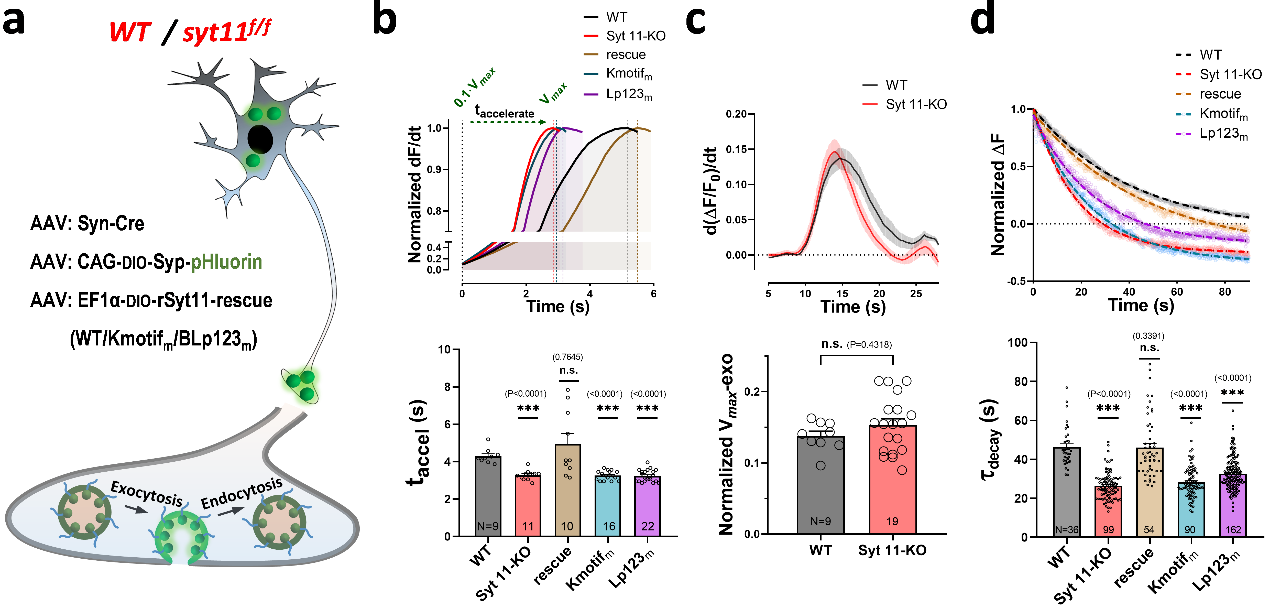
Syt11 阶段性动态调控神经兴奋-分泌偶联
为验证Syt11对突触囊泡胞吐-胞吞的动态调控,研究团队通过小鼠原代皮层神经元突触的Syn-pHluorin荧光成像发现,Syt11敲除(KO)后,胞吐的加速阶段(从胞吐开始到最大胞吐速率)时间缩短,而K-motif及loop-123突变体无法回补KO效果,表明Syt11可抑制早期胞吐事件,且此作用依赖于其膜结合能力;同时,Syt11 KO并不影响最大胞吐速率,表明此阶段(胞内Ca2+浓度达最大值)Syt11抑制解除、并不参与其胞吐调控;有趣的是,Syt11 KO还显著加速胞吐后的囊泡内吞过程,表明Ca2+对Syt11的抑制作用解除,Syt11与质膜的互作得以恢复,进而抑制囊泡胞吞过程。这些结果表明,Syt11对囊泡胞吐-胞吞的调控是阶段性的、受胞内Ca2+水平实时动态调控,进而建立了Ca2+依赖性的Syt11-Syt1质膜结合动态切换模型,揭示神经元兴奋-分泌动态偶联的分子基础与调控机制。
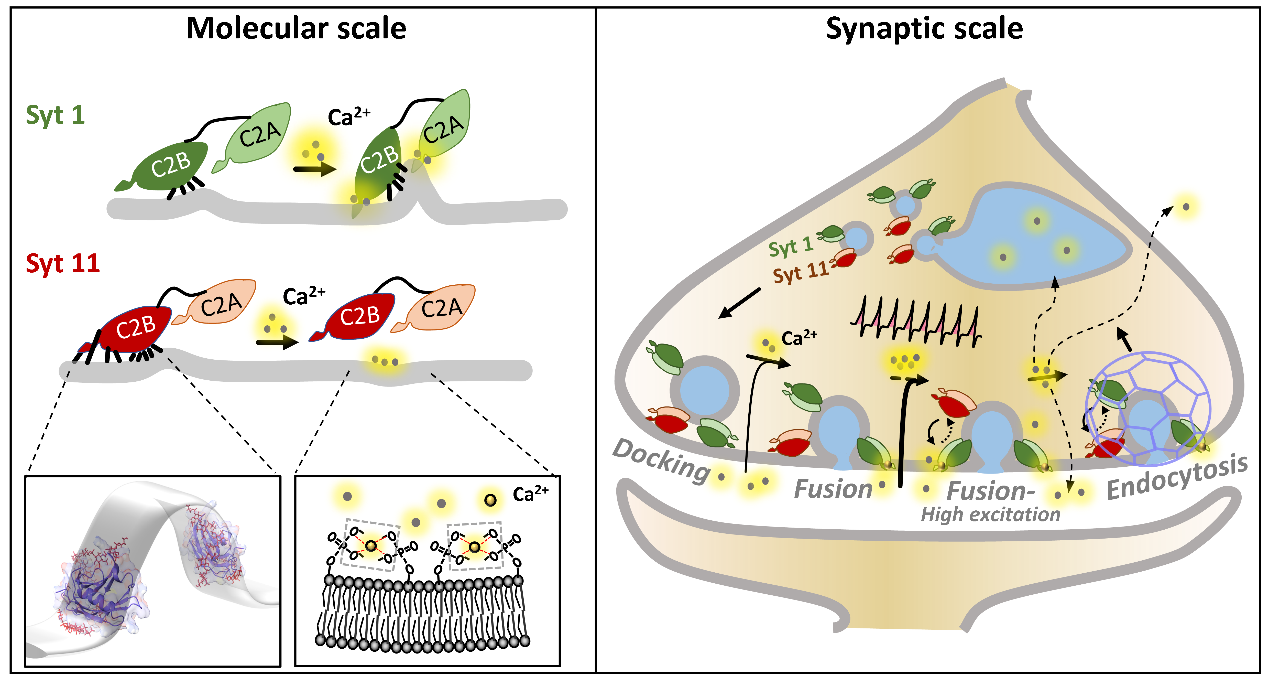
Syts的Ca2+敏感性、膜结合模式及膜占位切换模型
该研究利用生化分析、荧光成像等特色技术手段,发现了Syt11的 Ca2+抑制性质膜结合模式,并首次提出了神经突触分泌过程中Syt11与Syt1膜占位动态切换理论模型:静息状态下,囊泡上的Syt11与Syt1竞争性结合突触前膜而促进囊泡锚定(docking);动作电位到达后,Syt1响应低浓度Ca2+而触发膜融合,Syt11因无法响应Ca2+而抑制融合;在高强度或持续性兴奋时Ca2+浓度迅速升高,促进Syt1与质膜结合,同时抑制Syt11的竞争性结合,Syt1膜占位增加,囊泡融合迅速扩张;兴奋结束后Ca2+浓度迅速回落,Syt11恢复竞争力而重新占据膜表面,进而抑制胞吞后的胞吞过程,最终实现神经兴奋依赖性的胞吐-胞吞动态平衡与精准调控。
该研究工作首次揭示Syt11通过钙敏感的膜结合行为参与神经分泌调控,突破了“只有钙结合型Syt才受钙调控”的传统认知,阐明了Syt1与Syt11分别与质膜Ca2+敏感性结合的分子基础,提出了“CaR-Syt与CaT-Syt膜占位切换”理论模型,不仅为突触传递中 “兴奋-分泌”偶联和“胞吐-胞吞”偶联提供了新理论,还为理解神经分泌异常相关疾病的病理机制提供了新视角与新思路。

12月16日,该研究成果以《钙敏感型突触结合蛋白11与脂质的相互作用调节胞吐-胞吞过程》(Calcium-sensitive synaptotagmin 11-lipid interaction modulates exo-endocytosis)为题发表在《自然通讯》(Nature Communications)上。西安交通大学生命学院及生物医学信息工程教育部重点实验室为该论文的第一作者单位和通讯作者单位。生命学院徐华栋副教授、王昌河教授、宋茜副教授、基础医学院焦联营教授为该文共同通讯研究团队。生命学院博士生吴轩昂、姚靖宇、霍靖骁为论文共同第一研究团队。西安交通大学分析测试中心工程师马亚云、甘明明协助了ELS数据的采集。西安交通大学博士毕业生胡亚冲、刘彦玲博士为本工作提供了宝贵意见与帮助。本研究得到国家自然科学基金、陕西省自然科学基金、三秦英才特殊支持计划创新创业团队、陕西省重点研发计划、陕西省博士后基金等项目资助。
原文链接:https://www.nature.com/articles/s41467-025-67320-4








