近日,西安交大人工智能与机器人研究所暨人工智能学院博士生张贺与微软亚洲研究院AI for Science团队联合提出了基于人工智能技术和无轨道密度泛函理论的新型电子结构计算框架(M-OFDFT),不仅显著超越了传统KSDFT的计算效率,还能保有其求解精度,为分子科学研究提供了一种新颖的、更具潜力的研究工具。相关研究成果“Overcoming the Barrier of Orbital-Free Density Functional Theory for Molecular Systems Using Deep Learning在Nature子刊《自然计算科学》(Nature Computational Science)在线发表。
分子体系是生物、化学和材料科学等众多基础学科的基本研究对象,而电子结构方法是求解分子体系各种物理化学性质的基础工具。Kohn-Sham密度泛函理论(KSDFT)因其在计算精度和效率方面良好的权衡,成为目前分子科学研究中应用最为广泛的电子结构方法之一,但较高的计算复杂度影响了KSDFT在大规模分子体系上的应用。相较之下,无轨道密度泛函理论(OFDFT )的计算复杂度更低,但因缺乏适用于分子体系动能密度泛函,其在实际应用中的求解精度受到了很多限制。
为此,人机所博士生张贺等人通过引入一个高精度的分子动能密度泛函模型,提出了一种名为M-OFDFT的深度学习框架,在常见分子体系上能够用更小的时间代价求解出KSDFT级别精度的电子状态及分子属性。实验结果显示,相比利用神经网络“端到端”直接预测分子属性,M-OFDFT在远超训练集分子规模的体系中展现出了更好的外推性能。该成果也展示了利用AI技术推进电子结构方法计算精度和效率权衡的可能性,为加速科学发现注入了新的活力。

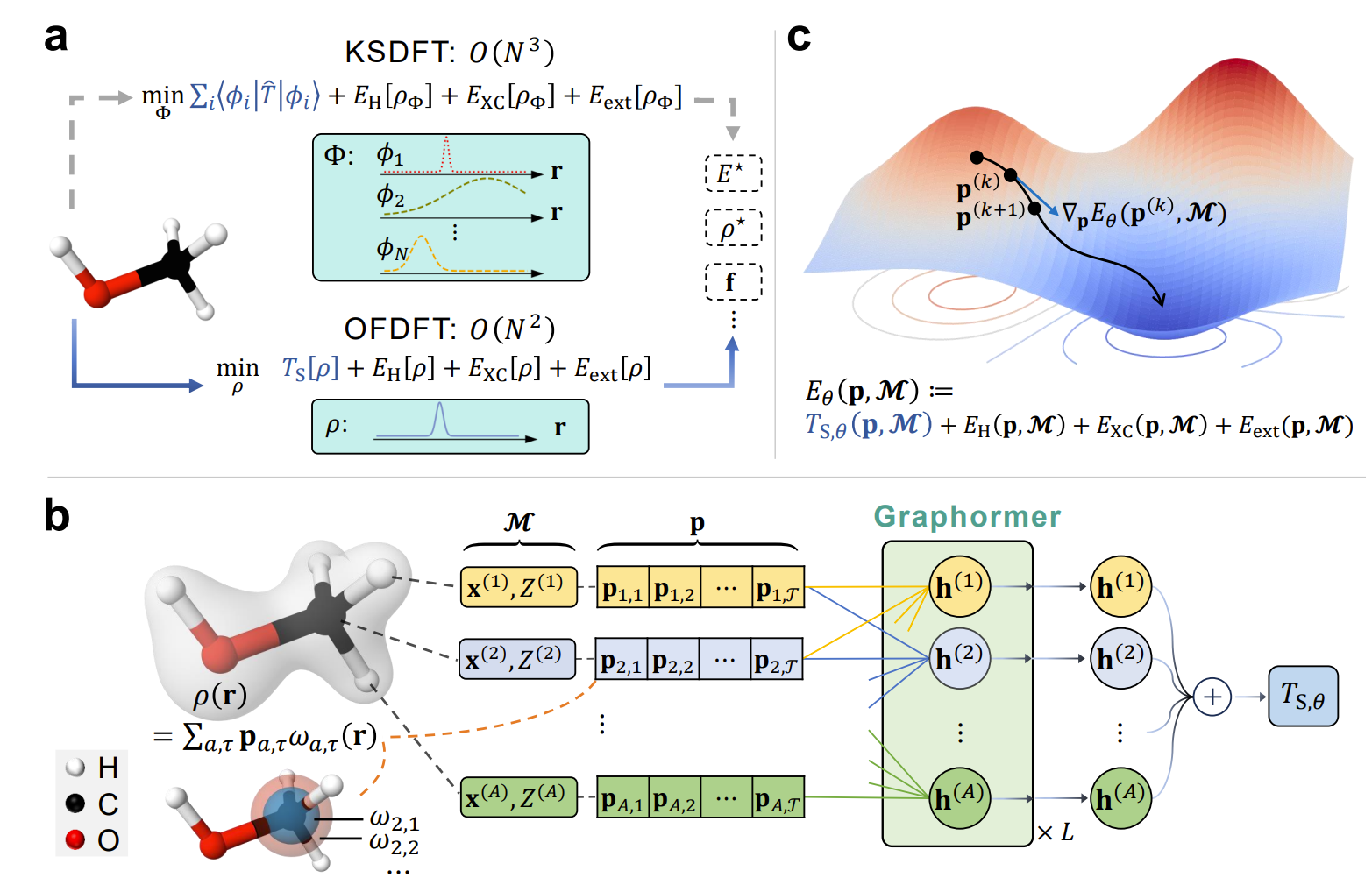
图1. M-OFDFT框架概览(a) 相比较KSDFT,无轨道密度泛函理论(OFDFT)理论复杂度更低; (b) 基于非局域图神经网络的动能密度泛函模型,该模型以电子密度系数和分子构象作为输入预测分子的电子动能;(c) 动能泛函模型训练完成后,M-OFDFT通过不断优化电子密度系数求解基态电子密度及分子相关性质。
文章第一作者为人工智能学院与微软亚洲研究院联合培养的博士生张贺,师从郑南宁教授和邵斌研究员,第一作者单位为西安交通大学。该研究工作是张贺于微软亚洲研究院刘畅研究员、邵斌研究员及郑南宁教授的联合指导下完成。
《自然-计算科学》文章链接:https://www.nature.com/articles/s43588-024-00605-8
SharedIt链接:https://rdcu.be/dANtS
论文链接:https://arxiv.org/abs/2309.16578








